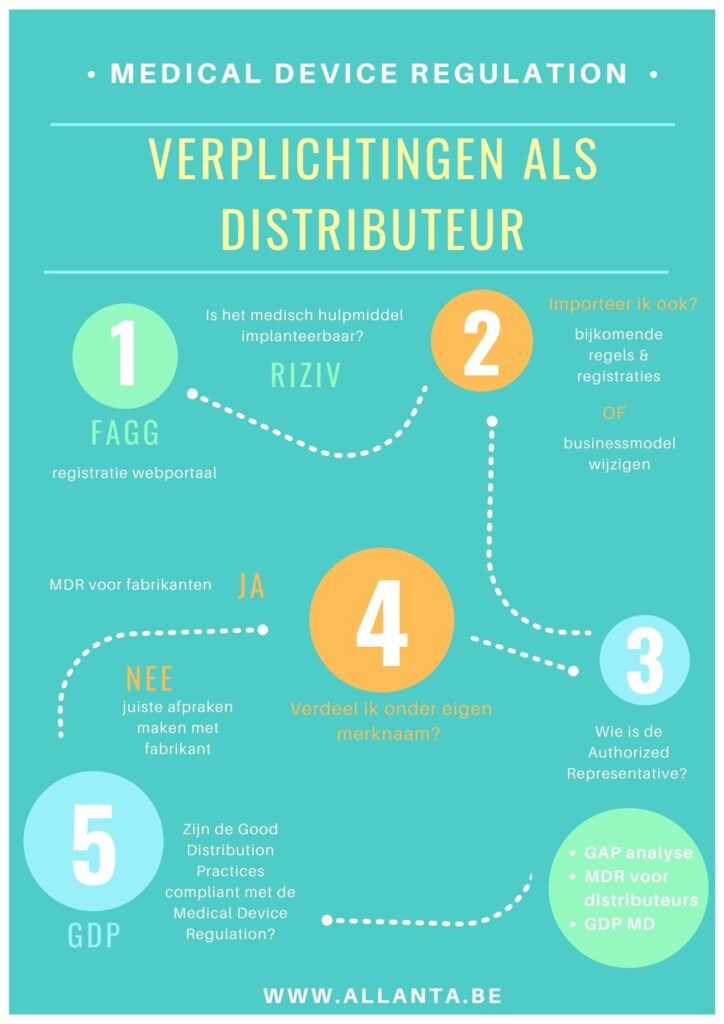
Wie medische hulpmiddelen wil verdelen in de Belgische markt, of binnen de Europese Unie mag er zeker van zijn dat je te maken krijgt met de Medical Device Regulation. Wat zijn jouw MDR verplichtingen als distributeur? In deze blog overlopen we vijf belangrijke onderdelen waarmee je moet rekening houden.

Om te weten wat jouw MDR verplichtingen zijn als distributeur, is het belangrijk eerst te bepalen wanneer je een distribiteur bent van medische hulpmiddelen. De MDR 2017/745 definieert het als volgt: elke natuurlijke of rechtspersoon in de toeleveringsketen, verschillend van de fabrikant of de invoerder, die een product op de markt van de Europese Unie aanbiedt tot het tijdstip van ingebruikneming. (Verordening EI 2017/745, artikel 2, 34 distributeur verordening).
Aangezien een distributeur mee betrokken is in de toeleveringsketen van medische hulpmiddelen is deze mee verantwoordelijk. Welke wettelijke eisen en richtlijnen nu specifiek voor welke actor (fabrikant, importeur, distributeur of gemachtigde) gelden, is niet altijd duidelijk voor distributeurs. Met veel foutieve handelingen en aanvragen tot gevolg die de in het markt brengen van medische hulpmiddel enorm kunnen vertragen. We geven je vijf verplichtingen mee waar je als distributeur van medische hulpmiddelen op moeten letten.
Verdelers (of distributeurs) met een maatschappelijke zetel in België of actief in de Belgische markt dienen zich te registreren via het webportaal van het Fagg.
Bepaal welke rollen je als organisatie opneemt. Is jouw activiteit enkel beperkt tot distributeur of is het medisch product afkomstig uit een land buiten de EU? Zo ja, dan treedt jouw bedrijf ook op als importeur. Goed over nadenken. want daar zijn extra verplichtingen en registraties aan verbonden. Of wijzig jouw businessmodel zodanig om de bijkomende rol als actor importeur te mijden.
De Europese wetgeving eist dat er een Authorized Representative (gemachtigde of EC Rep) is aangesteld. Deze EC Rep treedt op als contactpersoon en verantwoordelijke voor de autoriteiten. Zorg ervoor dat je weet wie de internationale fabrikant vertegenwoordigt voor de Europese markt.
Breng je het medische hulpmiddel onder jouw eigen merknaam in handel? Wordt het beoogde doeleind van een reeds in de handel gebracht of in gebruik genomen hulpmiddel gewijzigd? Of hebben de product wijzigingen gevolgen voor de naleving van de toepasselijke vereisten?
Als een van de antwoorden ja is, dan wordt jouw organisatie gezien als fabrikant. Er zijn vervolgens twee mogelijkheden.
Je stemt af met de oorspronkelijke ontwikkelaar van het product en maakt correcte afspraken zodat het FAGG duidelijk weet wie distribiteur en fabrikant is.
Of je neemt de rol op als fabrikant, registreert je organisatie als fabrikant, legt de CE technische documentatie voor aan de autoriteiten en laat je kwaliteitssysteem certificeren zodat je in regel bent met de Medical Device Regulation. Meer over het thema MDR en de weg naar certificatie lees je hier.
Good Distribution Practice (GDP) is een kwaliteitssysteem waarmee een distributeur de productkwaliteit en -veiligheid verzekert. Dit is een vereenvoudigde versie van een ISO 13485-kwaliteitssysteem en is gericht specifiek gericht op de rol als distributeur. In welke mate is het huidig GDP-kwaliteitssysteem afgestemd aan de wettelijke eisen voor distributeurs rekening houdend met de MDR? Of is er een opfrissing nodig? Hoe begin je er aan? In deze blog delen onze GDP-experten hun ervaringen.
Ik begeleid organisaties om haar processen in overeenstemming te brengen met normen, richtlijnen, best-pratices en/of wet-en regelgeving. Afgelopen jaren verdiepte ik me in de medische sector, de rol van stakeholders, wet-en regelgevingen (EU, US), certificerende instanties (notified bodies), kwaliteitsnormen/richtlijnen en de rol van sector vertegenwoordigers. Lees meer

